
More Information
Submitted: 18 July 2019 | Approved: 26 July 2019 | Published: 27 July 2019
How to cite this article: Kechida M, Yahia WB, Mnari W. A fatal portal vein thrombosis: A case report. Arch Vas Med. 2019; 3: 007-008.
DOI: 10.29328/journal.avm.1001009
Copyright License: © 2019 Kechida M, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Hemoglobinuria; Paroxysmal; Venous Thrombosis; Pancytopenia; Anemia; Hemolytic
Abbreviations: PNH: Paroxysmal Nocturnal Hemoglobinuria; CT: Computerized Tomography; PIG-A: Phosphatidyl Inositol Glycan; LDH: Lactate Dehydrogenase; FLAER: Fluorescein- Labeled Proaerolysin; GPI-AP: Glycosylphosphatidyl Inositol Anchored Proteins

A fatal portal vein thrombosis: A case report
M Kechida1*, W Ben Yahia1 and W Mnari2
1Internal Medicine and Endocrinology Department, Fattouma Bourguiba University Hospital, Monastir, Tunisia
2Radiology Department, Fattouma Bourguiba University Hospital, Monastir, Tunisia
*Address for Correspondence: M Kechida, Internal and Endocrinology Department, Fattouma Bourguiba University Hospital, Monastir, Tunisia, Tel: 0098 21697292219; Email: [email protected]
Background: Paroxysmal nocturnal hemoglobinuria (PNH) is a rare acquired hematologic condition which could be revealed by deep venous thrombosis. It could be fatal unless correctly treated.
Case report: We report here the case of a 28 year-old male with no medical history who was admitted to the emergency room for severe abdominal pain. Computerized Tomography angiography (CT) scan revealed portal vein thrombosis. Laboratory findings showed pancytopenia with severe regenerative normocytic anemia resulting in PNH. Because of the lack of Eculizumab, treatment was first based on curative anticoagulation until bone marrow transplant, with no success.
Conclusion: PNH remains a severe disease with bad prognosis unless treated with Eculizumab.
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired hematopoietic disorder characterized by non autoimmune hemolytic anemia, thrombotic manifestations and bone marrow failure. It’s caused by somatic mutation of the X linked phosphatidyl inositol glycan (PIG-A) gene resulting in an increased susceptibility to complement mediated lysis cells. Treatment is based on Eculizumab, a fully humanized immunoglobulin monoclonal antibody to C5 fraction of the complement which inhibit terminal complement activation and membrane attack complex formation resulting in hemolysis. PNH is still a threatening condition especially when it’s revealed by thrombosis, in countries where Eculizumad is not available. We report the case of a young man with PNH revealed by portal vein thrombosis to emphasize on such a complication during PNH and to discuss its management through literature review.
A 28 year-old male with no medical history was admitted to the emergency room for severe abdominal pain evolving for 3 days. On examination he was afebrile with marked pallor, scleral icterus, abdominal distension and diffuse epigastric tenderness associated with splenomegaly. Laboratory investigations showed pancytopenia with severe regenerative normocytic anemia, hemoglobin at 5.5 g/dl, platelets at 70 000 elements/ mm3 and white blood cells at 3300 elements/ mm3. Serum bilirubin was raised with predominant indirect bilirubin component associated to elevated Lactate dehydrogenase (LDH) level at 1600 UI/l. Direct antiglobulin test was negative. Renal function and electrolytes were into normal range. A CT scan was performed showing acute right and left portal vein thrombosis (Figure A) associated to a thrombosis of left collateral branches of the superior mesenteric vein (curved arrow, Figure B) and a thickened necrosis small bowel loop (large arrow, Figure B). Flow cytometry showed the substantial loss of 95% of expression of antigens CD66 and CD16 in granulocytes, CD14 in monocytes, and Fluorescein- Labeled proaerolysin (FLAER) in granulocytes and monocytes. Other causes of thrombosis or hemolytic anemia were ruled out. All tests for infectious, systemic inflammatory diseases, and vasculitis remained negative especially antiphospholipid antibodies. Portal vein thrombosis revealing paroxysmal nocturnal hemoglobinuria was diagnosed. Anticoagulation therapy using 100 UI/kg/day low molecular weight heparin was started associated with folic acid and blood transfusion. Treatment with monoclonal antibody Eculizumab could not be used because of its non-availability. Procedure for bone marrow transplantation was started but the patient died 7 days later because of massive acute pulmonary embolism.
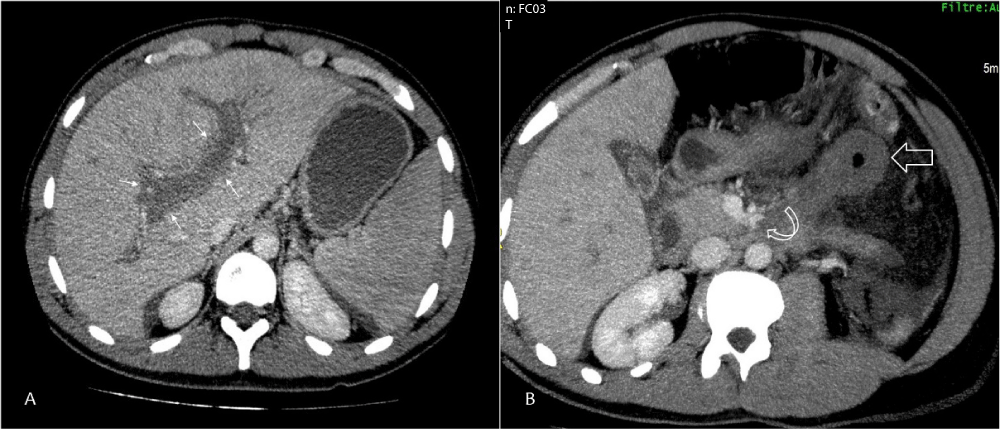
Figure 1: Portal vein thrombosis. Abdominal CT scan on venous phase contrast showed an acute right and left portal vein thrombosis (small’s arrows, Panel A). MIP and multiplanar reconstructions revealed a thrombosis of left collateral branches of the superior mesenteric vein (curved arrow, Panel B) and a thickened necrosis small bowel loop (large arrow, Panel B).
Paroxysmal nocturnal hemoglobinuria is a clonal stem cell disorder that is caused by somatic mutation of the X-linked phosphatidyl-inositol glycan (PIG-A) gene, resulting in partial or absolute deficiency of all glycosylphosphatidyl inositol anchored proteins (GPI-AP), as PIG-A is involved in the initial stage of synthesis of the GPI anchor (1). Clonal cells with deficiency of GPI anchored proteins can be erythrocytes, platelets, polymorphonuclear leukocytes, monocytes and B cells, T cells and natural killer lymphocytes (1). This loss of glycosylphosphatidyl inositol anchor protein mainly CD55 and CD59 (1) results in an increased susceptibility to complement mediated lysis of these cells which will be responsible for clinical manifestations: hemolytic anemia, pancytopenia due to bone marrow failure and thrombosis.
Incidence of thrombosis in PNH can reach up to 10% of patients revealing the disease but cumulative incidence over 8 to 10 years can reach 23% to 30% (2). Venous thrombosis are more common than arterial ones [1,2]. The most frequent localization is hepatic veins (40%) (2) leading to Budd Chiari Syndrome but other abdominal sites like portal, mesenteric and splenic thrombosis are less frequently affected in addition to cerebral thrombosis [1].
Mechanisms leading to thrombosis are still unclear [1]. It’s suggested that CD59 deficient platelets are activated by complement leading to a procoagulant activity with thrombus generation (1). A part from that, the prothrombotic state may be determined by underlying hemolysis and the impairment of the fibrinolytic system associated to complement mediated endothelial cell damage [3].
Thrombosis in PNH is accounting for 50% of deaths without treatment [4]. Management of thrombosis in PNH is now based on Eculizumab. It is a fully humanized immunoglobulin monoclonal antibody to C5 which inhibit terminal complement activation and membrane attack complex formation that results in hemolysis [2]. In addition to that full dose anticoagulation is mandatory unless no contraindications. Anticoagulation alone is not effective as thrombosis will be extended like in our patient. In countries where eculizumab is not available, bone marrow transplant may be an alternative curative option [2]. That was discussed in our patient even if that was associated to an overall survival probability of 54% [5]. Unfortunately there were no sufficient time to do that and the patient died because of thrombosis extension and pulmonary embolism despite optimal anticoagulation.
MK is the main author of the article, WBY contributed in writing the manuscript, WM: performed CT scan and interpreted the radiological findings. All authors read and approved the final manuscript.
- Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993; 73: 703-711. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8500164
- Morag Griffin and Talha Munir. Management of thrombosis inparoxysmal nocturnal hemoglobinuria: a clinician’s guide. Ther Adv Hematol. 2017; 8: 119-126.PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28246555
- Ageno W, Dentali F, De Stefano V, Barco S, Lerede T, et al. Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria phenotype inpatients with splanchnic vein thrombosis. Thromb Res. 2014; 133: 1052-1055. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24731559
- Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood. 2013; 121: 4985-4996. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23610373
- de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008; 112: 3099-3106. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18535202